
Solution | Molecular Property Prediction#
Overview#
Business case#
Exploring new drugs to improve human health is a significant effort that requires collaboration among scientists, clinicians, and regulatory authorities to bring new treatments to patients.
The process involves identifying a target protein related to a specific disease, finding or designing molecules that interact with that protein in a desired way, and testing these molecules rigorously. Finally, if successful, this process requires developing these molecules into safe and effective drugs for treating the disease. It’s a complex and time-consuming process that can cost around $2.6 billion and take about 12 years on average.
In recent years, this field has undergone a substantial transformation through the integration of AI. Notably, the investment in AI-enabled drug development has experienced a remarkable surge. It reached $59.3 billion as of 2023, a nearly 27-fold increase since 2015 (Source: Deep Pharma Intelligence).
Analysts project that a 20-40% reduction in preclinical development costs could provide the financial resources required to advance four to eight novel molecules successfully. Biotech companies embracing an AI-driven approach have cultivated an impressive pipeline of potential drugs. Boasting over 150 small-molecule drugs in the discovery phase and 15 undergoing clinical trials, the role of AI in drug discovery becomes undeniable.
This Dataiku Molecular Property Prediction Solution aims to optimize the process of molecular screening on selected target proteins by querying molecules with known bioactivity via ChEMBL and PubChem databases.
The Solution enables the deployment of machine learning models to predict molecular properties from chemical structures. These models can guide the digital drug discovery process by identifying the most promising drug candidates before anyone conducts experimental work.
Installation#
From the Design homepage of a Dataiku instance connected to the internet, click + Dataiku Solutions.
Search for and select Molecular Property Prediction.
If needed, change the folder into which the Solution will be installed, and click Install.
Follow the modal to either install the technical prerequisites below or request an admin to do it for you.
Note
Alternatively, download the Solution’s .zip project file, and import it to your Dataiku instance as a new project.
Technical requirements#
To use this Solution, you must meet the following requirements:
Have access to a Dataiku 13.4+* instance.
Accessing ChEMBL and PubChem through an API service is currently not required.
A Python 3.9 code environment named
solution_molecular-prop-predictionwith the following packages:
Flask==3.0.2
PubChemPy==1.0.4
chembl-webresource-client==0.10.8
datamol==0.11.2
huggingface-hub==0.16.4
molfeat==0.9.2
python-dotenv==1.0.0
rdkit==2023.3.3
requests==2.31.0
tqdm==4.66.1
transformers==4.32.0
deepchem==2.7.1
The code environment also requires an initiation script that initializes a tokenizer and model from the Hugging Face library and manages permissions for cache directory access. Users should put the following script in the tab Resources.
## Base imports
from dataiku.code_env_resources import clear_all_env_vars
from dataiku.code_env_resources import set_env_path
from dataiku.code_env_resources import grant_permissions
from transformers import AutoTokenizer, AutoModel
# Clears all environment variables defined by previously run script
clear_all_env_vars()
## Hugging Face
# Set HuggingFace cache directory
set_env_path("TRANSFORMERS_CACHE", "huggingface")
tokenizer = AutoTokenizer.from_pretrained("DeepChem/ChemBERTa-77M-MLM", cache_dir="huggingface")
model = AutoModel.from_pretrained("DeepChem/ChemBERTa-77M-MLM", cache_dir="huggingface")
grant_permissions("huggingface")
The downloadable version uses filesystem-managed datasets and the built-in Dataiku engine as the only processing engine. Performance could be significantly improved by changing all the connections to Snowflake connections.
Data requirements#
Note
This Solution uses data pulled via the ChEMBL and PubChem API but isn’t endorsed or certified by these organizations. By utilizing this Solution, you agree to abide by the terms set forth on these data sources.
The Solution directly queries the ChEMBL or PubChem API based on user specifications. It loads the results into metadata, which captures an overview of the user query and stores all the molecules studied before and reported on the database. The current API version doesn’t require API keys for the connection. The Solution automatically applies all the required preprocessing to store the data in the output schema.
The pre-uploaded ClinTox dataset is a benchmark dataset from MoleculeNet, designed to predict drug toxicity based on chemical structure. It contains 1,491 drug compounds with known toxicity profiles compiled from FDA approvals and clinical trial failures.
Input data from the user for scoring novel molecules. The user must upload their test_data for scoring novel molecules with the structure below.

Workflow overview#
You can follow along with the sample project in the Dataiku gallery.
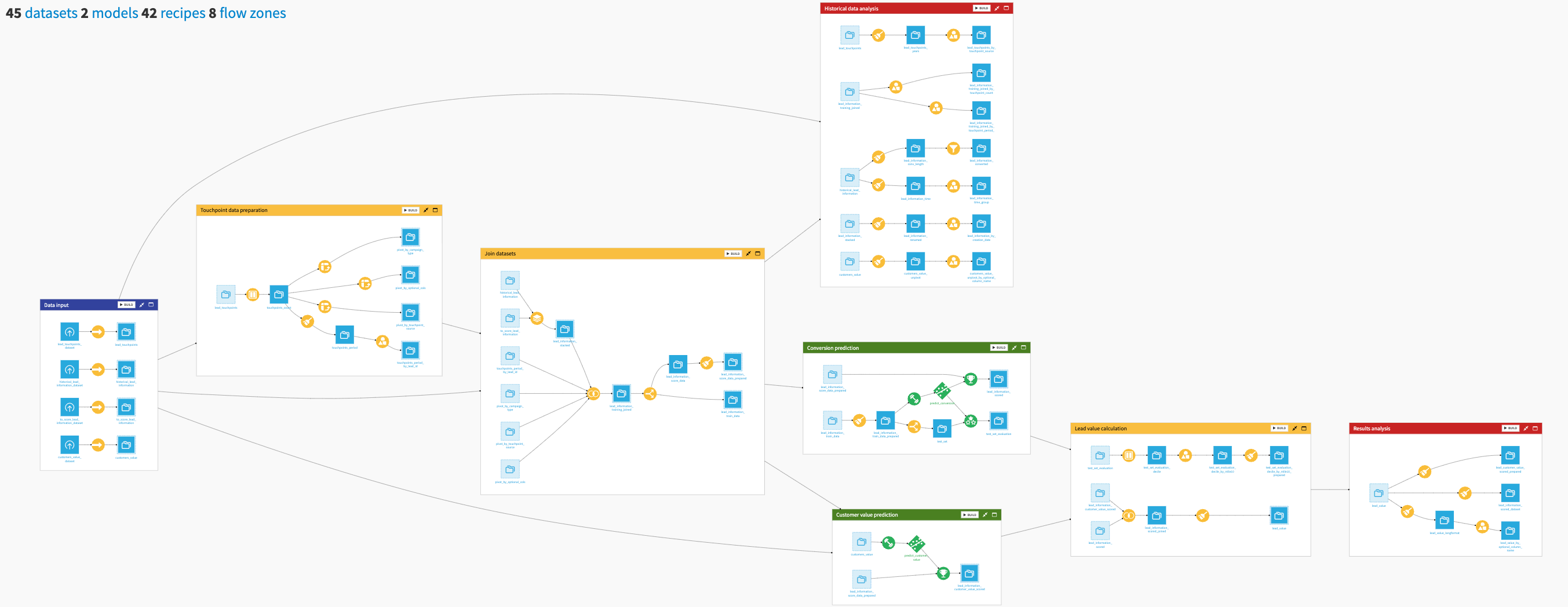
The project has the following high-level steps:
Query molecules (SMILES) with known bioactivity for a specific protein target via ChEMBL or PubChem API to predict bioactivity.
Generate molecular descriptors and fingerprints to perform quantitative structure-activity analysis and understand how molecular properties influence bioactivity.
Train and benchmark machine learning models to predict molecular bioactivity (pIC50) as a measure of molecule potency and molecular toxicity for safety to speed up experimental work on large datasets.
Score novel molecules and prioritize the ones that qualify for the next discovery stage under the required properties.
Assess further compound similarity using t-SNE and statistics that help to identify structurally related studied compounds to validate potential drug targets.
Publish the results to a template dashboard that showcases the analysis, modeling, and novel molecule output.
Walkthrough#
Note
In addition to reading this document, it’s recommended to read the wiki of the project before beginning to get a deeper technical understanding of how this Solution was created and more detailed explanations of Solution-specific vocabulary.
Solution setup via Dataiku app#
The Molecular Property Prediction Dataiku app helps configure the project’s critical parameters. It subsequently builds the elements in the Flow zones. It also enables multiple users to work on individual instances of the Solution without directly modifying the original project.
The first part of the app validates that the protein accession code exists in the chosen public database. The second part consists of an interface for the user to enter the project variable selection for the required analysis. The next step is to replace the test_data and use the scenario BUILD ALL to build the Flow and update the dashboard.
Below are explanations of the different variables that users need to manually set in the project. You can modify them in the Variables section of the project.
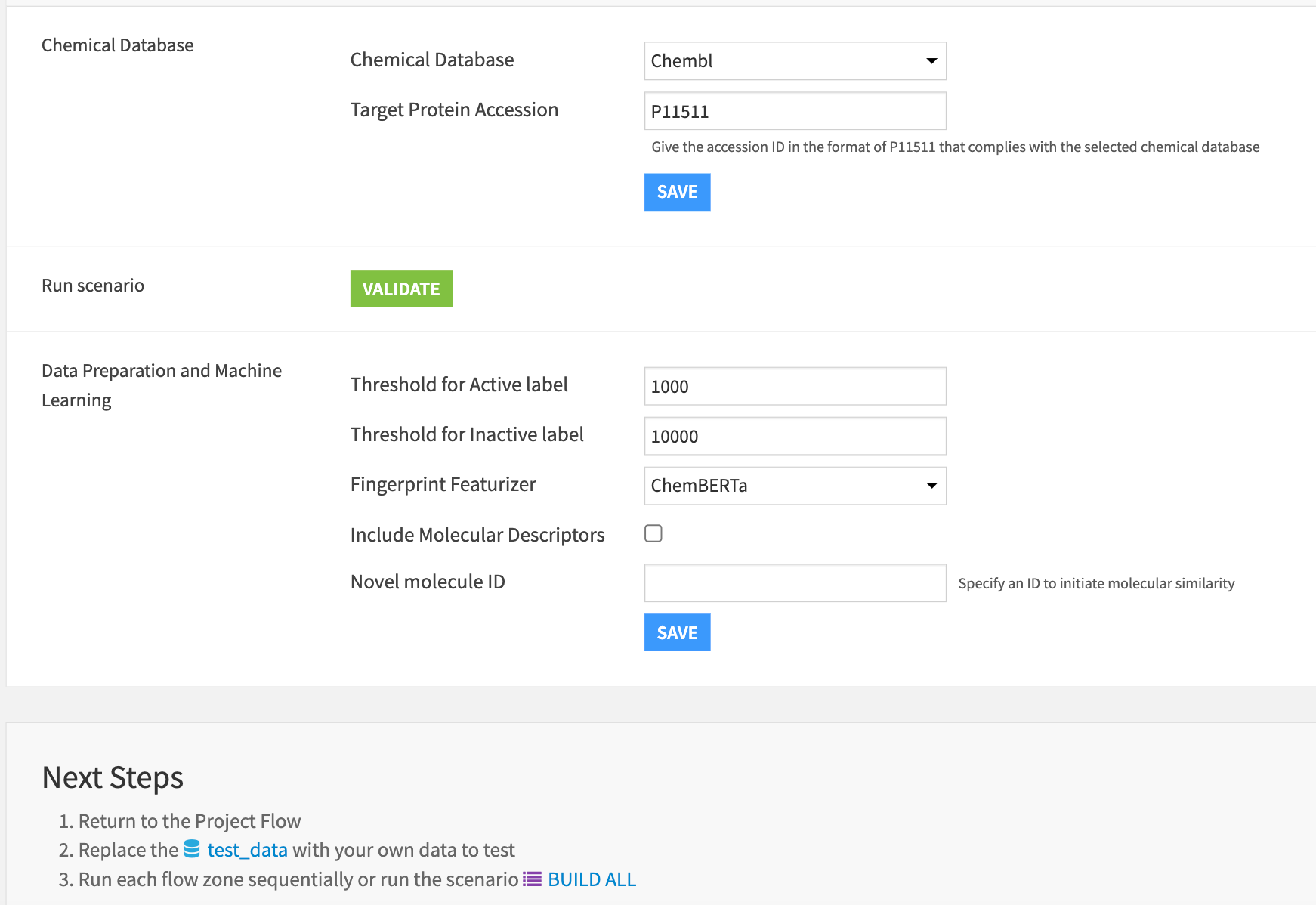
Database selection#
Select a public chemical database to connect automatically through API and specify the target protein accession code. The first scenario validates the presence of the accession code in the selected database.
Data preparation#
Specify the parameters required for data preparation.
Molecular bioactivity is defined as a molecule’s ability to bind to a biological target based on the standard value IC50.
A molecule is defined as Active if the IC50 value < Threshold for Active label.
A molecule is defined as Inactive if the IC50 value > Threshold for Inactive label.
Otherwise intermediation.
Machine learning#
Descriptors are quantitative metrics that characterize the chemical and physical properties of molecules. These descriptors comprise the input features to the regression model for predicting the molecular bioactivity value pIC50.
Molecular descriptors |
Capture physiochemical properties as continuous numerical values. Examples include molecular weight and number of atoms. |
Fingerprint descriptors |
Represent the presence or absence of specific chemical features as binary or numerical codes generated from the canonical smile notations. Often used for similarity searches and clustering of compounds in chemical databases. Examples include Morgan Fingerprint (ECFP4 analog, 1024-bit-long), MACCS keys, PubChem fingerprint, and large-scaled pre-trained model ChemBERTa from hugging face. |
By default, the machine learning models use the fingerprint descriptors only as input features. The user has the option below to include both.
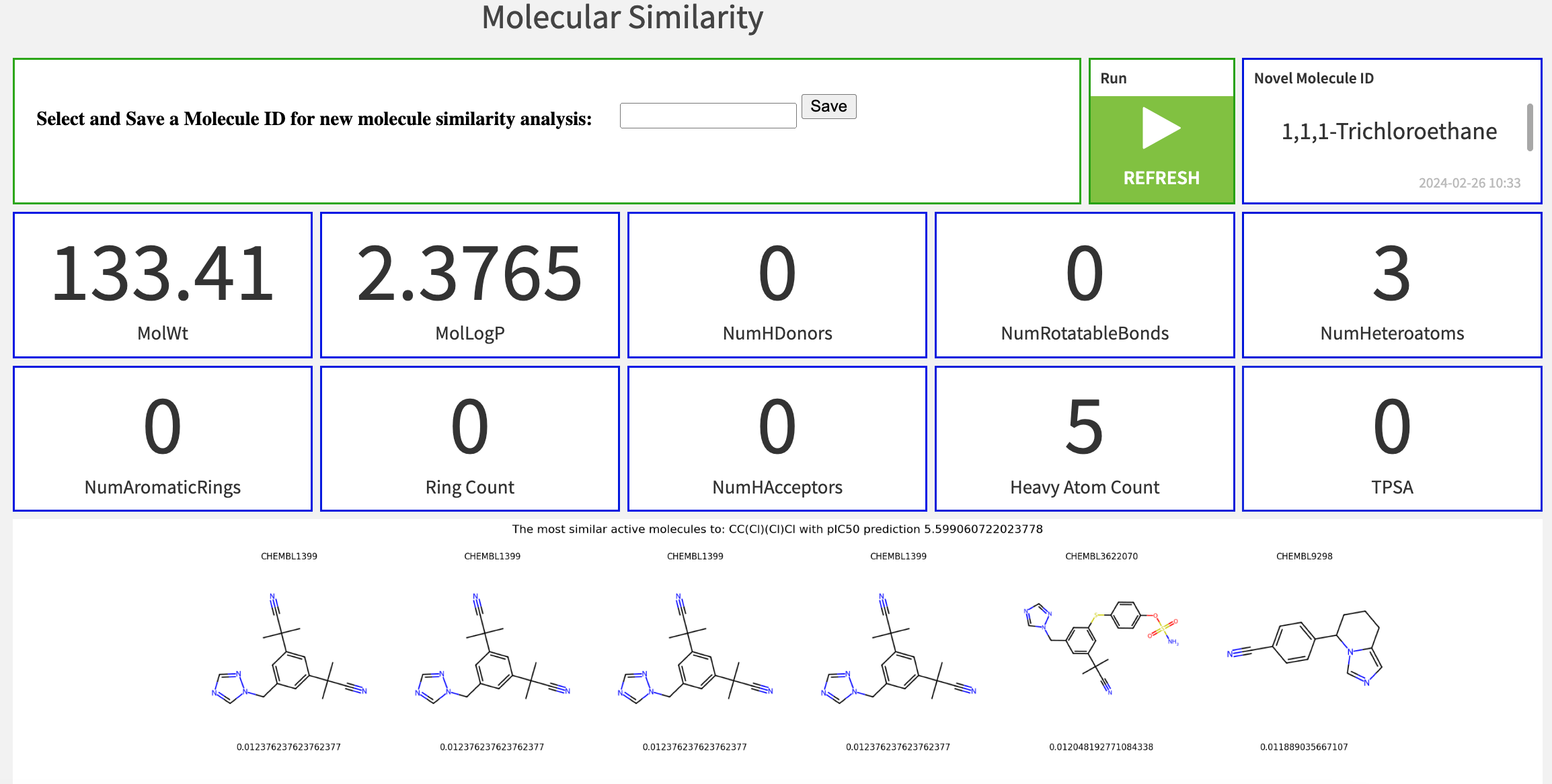
Molecular similarity#
The project analyzes the degree of structural resemblance or likeness between novel scored molecules and studied molecules used for training. The final field of the Dataiku app allows you to specify a novel molecule ID to initiate the analysis. You can dynamically interact with all the novel molecules within the dashboard results.
Explore the chemical space of studied molecules and their influence on the target protein#
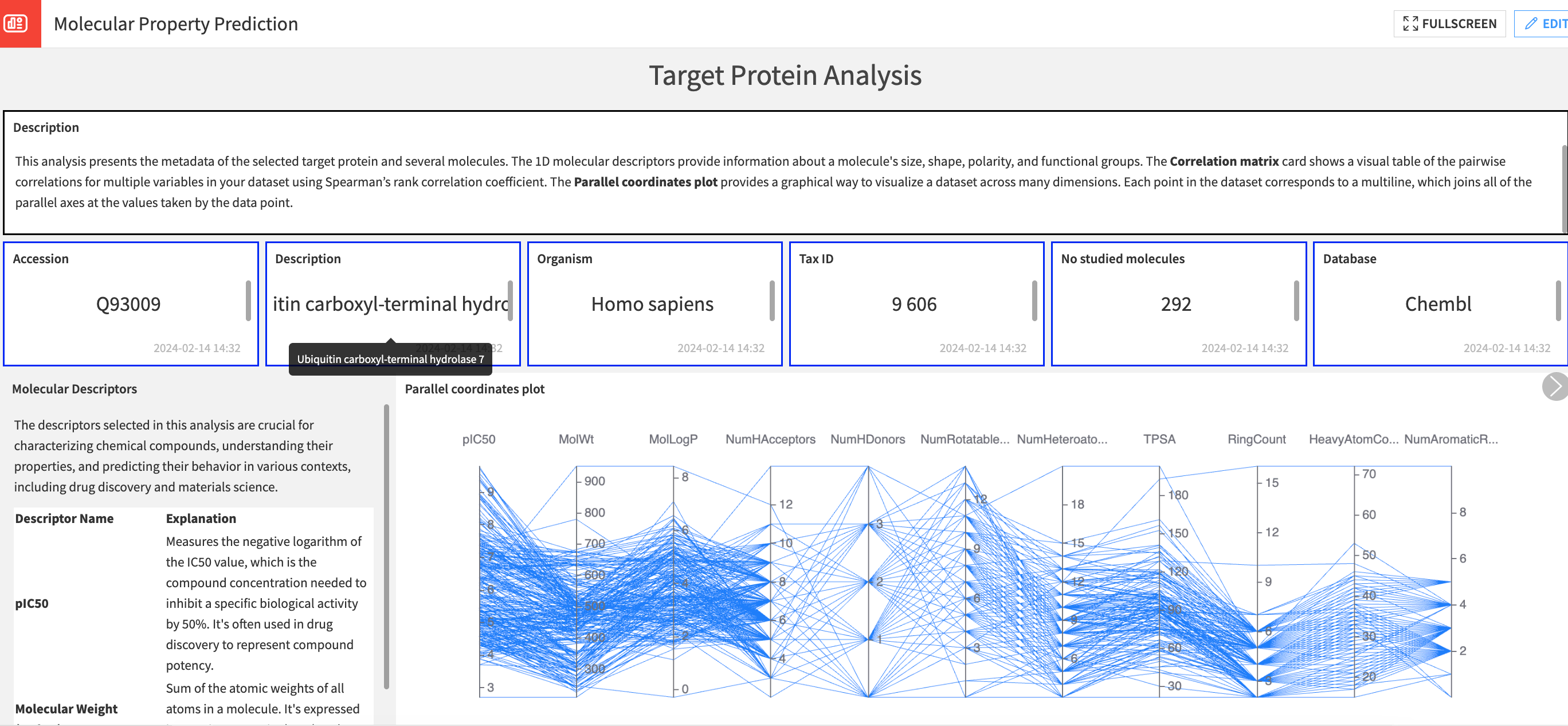
The target protein analysis presents the metadata of the selected target protein and several molecules. The 1D molecular descriptors provide information about a molecule’s size, shape, polarity, and functional groups.
The chemical space analysis allows the users to visualize, explore, and analyze the relationships between molecular structures and their properties.
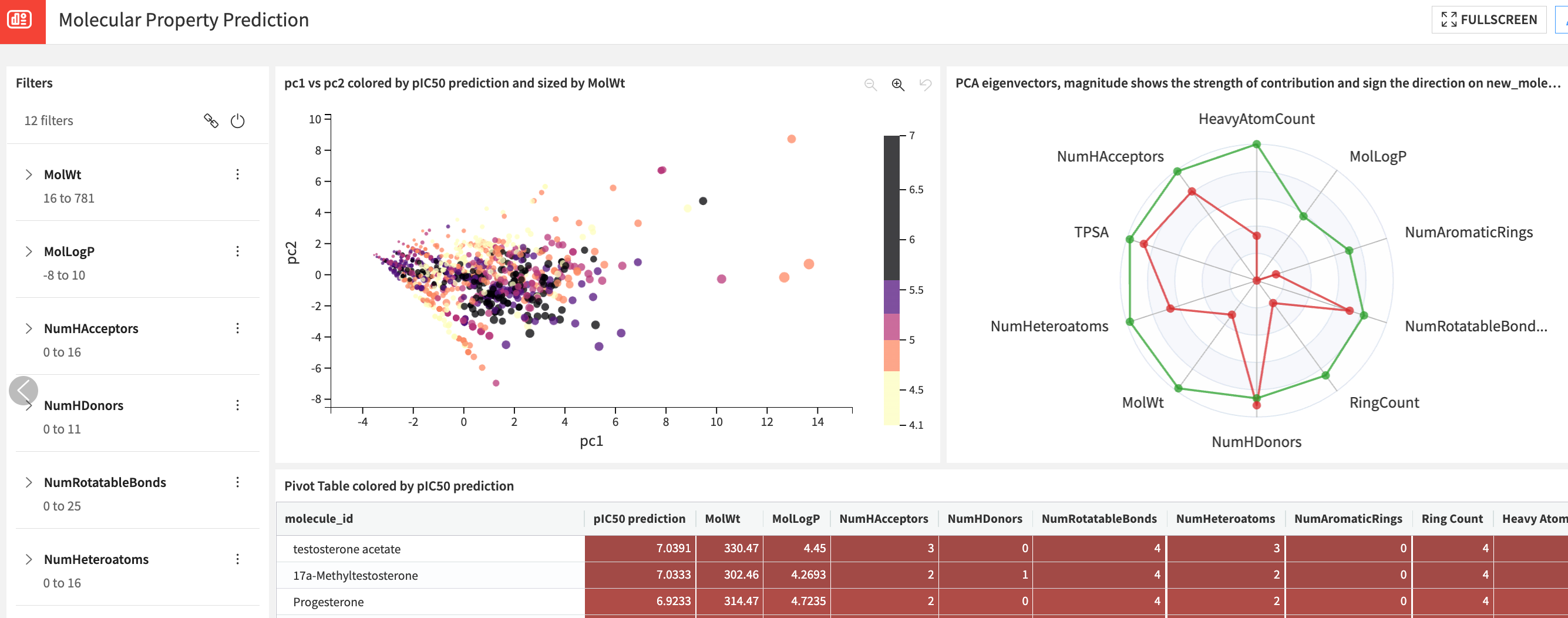
Discover novel molecules#
Machine learning models help prioritize compounds for experimental testing by identifying those more likely to be effective against a specific target and also less harmful (toxic). Combining both the pIC50 prediction, toxicity and the molecular descriptors from RDkit, you can filter down to your search space of interest and prioritize the compounds to pursue further.
This process is especially valuable in the early stages of drug discovery when resources are limited and researchers need to make informed decisions. This exploration can uncover novel compounds with unique structural features with therapeutic potential.
Identify molecules with similar structures#
Molecular similarity is the degree of likeness between two molecules based on their structural properties. The molecular similarity score is computed with the Tanimoto Coefficient, which compares the presence or absence of structural features (for example, molecular fingerprints) between pairs of new and studied molecules.
Assessing molecular similarity helps researchers identify structurally or functionally related molecules. This can have significant implications in use cases such as virtual screening for drug discovery, chemical toxicity prediction, and chemical library design.
Responsible AI statement#
We value responsible development and deployment of AI in drug discovery. While our pIC50 prediction model offers valuable insights into potential bioactivity, it’s crucial to remember that it’s only one piece of the puzzle.
Decisions affecting lab experiments shouldn’t solely rely on pIC50. Additional critical factors, such as ADMET properties (absorption, distribution, metabolism, excretion, and toxicity), selectivity (targeting the intended protein without harming others), and pharmacokinetic properties (drug movement and action in the body) must be rigorously assessed.
This model is designed to augment — not replace — the invaluable expertise of subject matter experts and the essential role of lab work. Combining in silico predictions with thorough biological validation and expert judgment can ensure responsible and ethical prioritization of experiments, ultimately accelerating the development of safe and effective drugs.
Reproduce these studies with minimal effort for your data#
This template Solution equips computational labs to speed up their work in the early stages of drug discovery by automating data analytics and the quantitative structure analysis relationship process. You can find a deeper technical walkthrough of the project within the wiki to aid in reproducing this project.
This documentation has provided several suggestions on how to derive value from this Solution. Ultimately however, the “best” approach will depend on your specific needs and data. If you’re interested in adapting this project to the specific goals and needs of your organization, Dataiku offers roll-out and customization services on demand.